
sindrome de apert
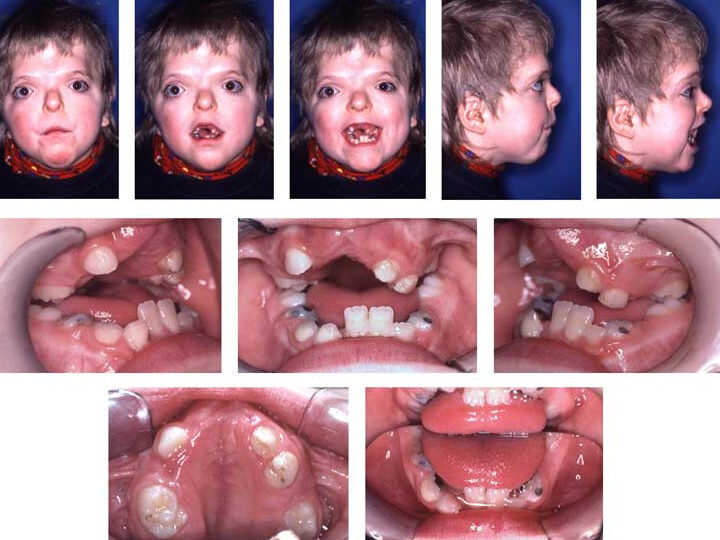
El síndrome de Apert, también conocido como síndrome de Apert o acrocefalosindactilia, es un trastorno genético raro caracterizado por la craneosinostosis, o la fusión prematura de los huesos del cráneo, lo que lleva a un crecimiento y desarrollo anormal de la cabeza y la cara. Esta condición afecta aproximadamente a 1 de cada 65,000 a 1 de cada 160,000 nacimientos en todo el mundo.
Causas del síndrome de Apert
El síndrome de Apert es causado principalmente por una mutación en el gen del receptor 2 del factor de crecimiento de los fibroblastos (FGFR2). Esta mutación genética interrumpe el desarrollo normal y afecta la formación de huesos y tejidos en el cráneo, las manos y los pies. La mutación ocurre al azar y no se hereda típicamente de los padres. Sin embargo, si un padre tiene el síndrome de Apert, hay un 50% de probabilidad de transmitirlo a sus hijos.
La investigación sugiere que la edad paterna avanzada en el momento de la concepción puede ser un factor de riesgo para la mutación. No se han identificado factores ambientales como contribuyentes significativos a la aparición del síndrome de Apert.
Síntomas del síndrome de Apert
El síntoma más común y notable del síndrome de Apert es la craneosinostosis, lo que resulta en una forma anormal del cráneo. Esto puede llevar a varias complicaciones, incluyendo un aumento de la presión en el cerebro, lo que puede causar retrasos intelectuales y del desarrollo.
Otros síntomas del síndrome de Apert incluyen:
1. Rasgos faciales anormales:
Las personas con síndrome de Apert a menudo tienen una apariencia facial distintiva, que incluye una frente alta y prominente; ojos prominentes y separados; una nariz en forma de pico; y una región media de la cara subdesarrollada. El crecimiento de la mandíbula superior puede estar restringido, lo que provoca problemas dentales y de mordida.
2. Fusión de dedos de las manos y de los pies:
Una de las características definitorias del síndrome de Apert es la sindactilia, que es la fusión o unión de los dedos de las manos y de los pies. Las manos y los pies pueden parecer como unos mitones, con los dedos fusionados. Esto puede afectar la destreza y obstaculizar las funciones normales de las manos y los pies.
3. Anomalías esqueléticas:
Las personas con síndrome de Apert pueden experimentar anomalías esqueléticas adicionales, como una curvatura anormal de la columna vertebral (escoliosis), fusión de ciertas vértebras o costillas faltantes o adicionales. Estos problemas esqueléticos pueden causar dolor y limitar la movilidad.
Tratamiento para el síndrome de Apert
Aunque no hay cura para el síndrome de Apert, el tratamiento se enfoca en manejar los síntomas y tratar las complicaciones asociadas. A menudo se requiere atención multidisciplinaria que involucra a diversos especialistas médicos para proporcionar tratamiento integral y apoyo.
Las opciones de tratamiento para el síndrome de Apert pueden incluir:
1. Cirugía:
Las intervenciones quirúrgicas son comúnmente necesarias para corregir la craneosinostosis y remodelar el cráneo. Estos procedimientos tienen como objetivo aliviar la presión en el cerebro, mejorar la estética y mejorar la función general de la cabeza y la cara.
2. Cirugías de manos y pies:
Para mejorar la función de las manos y los pies, se realizan procedimientos quirúrgicos para separar los dedos fusionados. Estas operaciones permiten una mayor destreza y mejoran la independencia individual.
3. Atención médica continua:
Los niños con síndrome de Apert requieren revisiones médicas regulares para monitorear su desarrollo, manejar complicaciones asociadas y brindar intervenciones adecuadas según sea necesario. Especialistas como cirujanos craneofaciales, neurocirujanos, ortodoncistas y genetistas colaboran para brindar atención integral.
4. Terapia del habla y ocupacional:
Las personas con síndrome de Apert pueden beneficiarse de terapia del habla y ocupacional para mejorar el desarrollo del habla, las técnicas de alimentación y las habilidades cognitivas. Estas terapias tienen como objetivo mejorar la calidad de vida en general y promover la independencia.
Vivir con el síndrome de Apert
Afrontar el síndrome de Apert puede ser desafiante tanto para los individuos como para sus familias. Sin embargo, con intervenciones tempranas, atención multidisciplinaria y redes de apoyo, las personas pueden llevar vidas plenas y alcanzar su máximo potencial.
Es importante que las familias se conecten con grupos de apoyo y organizaciones especializadas en el síndrome de Apert para recibir orientación, información y apoyo emocional.
En conclusión,
El síndrome de Apert es un trastorno genético raro caracterizado por la craneosinostosis, rasgos faciales anormales y fusión de dedos de las manos y de los pies. Aunque no hay cura, las opciones de tratamiento se centran en manejar los síntomas y las complicaciones asociadas. Con intervenciones adecuadas y apoyo, las personas con síndrome de Apert pueden llevar vidas significativas.
Recuerde consultar con profesionales de la salud para obtener un diagnóstico preciso, opciones de tratamiento y atención personalizada para las personas con el síndrome de Apert.
Deja una respuesta